La prise en compte de l'innovation thérapeutique dans les politiques de prix et de remboursement des médicaments
Une approche internationale
- Par Catherine Sermet
Pages 319 à 341
Citer cet article
- SERMET, Catherine,
- Sermet, Catherine.
- Sermet, C.
https://doi.org/10.3917/rfas.073.0319
Citer cet article
- Sermet, C.
- Sermet, Catherine.
- SERMET, Catherine,
https://doi.org/10.3917/rfas.073.0319
Notes
-
[*]
Directrice adjointe, IRDES, Institut de recherche et documentation en économie de la santé.
-
[1]
Source : Éco-Santé 2007 : http://www.ecosante.fr
-
[2]
http://www.afssaps.fr/php/ecodex/index.php, consulté le 21 septembre 2007.
- [3]
- [4]
-
[5]
Conclusion du Conseil du 29 juin 2000 sur les médicaments et la santé publique 2000/C 218/04, Journal officiel des Communautés européennes, 31 juillet 2000.
-
[6]
Résolution du Conseil du 2 décembre 2003, Médicaments et santé publique : les défis –
Priorité aux patients, Journal officiel, n° C 020 du 24 janvier 2004, p. 0002-0004. -
[7]
Règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004 établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments.
- [8]
-
[9]
Fiche technique de la Haute Autorité de santé : http://has.presstvnews.fr/recos2/docs/fiches-techniques-def/Fiche_technique_definitions.pdf
- [10]
- [11]
-
[12]
Par exemple, sont considérés comme comparables tous les solides administrés par voie orale, capsules, comprimés, granulés effervescents, etc. Ces formes pharmaceutiques comparables sont répertoriées en annexe du compendium des lignes directrices, politiques et procédures, document qui précise les procédures mises en œuvre par le Conseil d’examen des prix des médicaments brevetés : http://www.pmprb-cepmb.gc.ca/francais/view.asp?x=654.
-
[13]
Loi du 10 août 2001 portant des mesures en matière de soins de santé. http://www.staatsbladclip.be/lois/2001/09/01/loi-2001022579-Print.html
-
[14]
Arrêté royal du 21 décembre 2001, fixant les procédures, délais et conditions en matière d’intervention de l’assurance maladie obligatoire soins de santé et indemnités dans le coût des spécialités pharmaceutiques.
http://www.inami.fgov.be/drug/fr/drugs/reglementation/ar-20011221/pdf/arkb20011221.pdf - [15]
- [16]
- [17]
-
[18]
ATC : Anatomical, Therapeutical, Chemical ; cette classification s’applique aux substances actives et comporte cinq niveaux de hiérarchie. Une substance active peut avoir plusieurs codes ATC.
-
[19]
QALYs : Quality Ajusted Life Years, littéralement : années de vie ajustées par la qualité.
- [20]
1De 1990 à 2006, la dépense de médicament en France est passée de 18 à 32 milliards d’euros. En 2006, elle représente 20 % de la consommation médicale totale [1]. Plus de la moitié (50,6 %) de ces dépenses sont liées à des médicaments commercialisés depuis moins de dix ans et la part de marché des médicaments qui le sont depuis moins de cinq ans atteint 22,3 %. La contribution des médicaments de moins de dix ans à la croissance totale des ventes est de 8,7 points (Clerc, 2007). En 2006, 956 autorisations de mise sur le marché ont été délivrées et à ce jour 457 spécialités ont été commercialisées [2].
2Chacun de ces médicaments récents comporte une part variable d’innovation. La littérature sur l’innovation pharmaceutique distingue généralement la véritable innovation, qui ouvre la voie à une nouvelle classe thérapeutique ou au traitement d’une maladie grave, des médicaments dits « me-too », des molécules dérivées et des nouvelles présentations de substances anciennes. Les « me-too » sont des molécules appartenant à une classe existante, traitant la même maladie et qui ne diffèrent que très peu des molécules déjà sur le marché. Les molécules dérivées résultent de la modification minime de molécules existantes (isomères, sels, esters, métabolites actifs…), modifications visant à améliorer l’efficacité ou la tolérance du médicament. Enfin, des substances anciennes et existant sur le marché depuis plus longtemps peuvent également bénéficier de modifications de forme, de dosage ou de mode d’administration qui incluent la plupart du temps une part d’innovation.
3Seul un petit nombre de nouvelles substances, représentant des avancées thérapeutiques notables, est mis sur le marché chaque année. En 2006, la revue Prescrire ne compte que 23 nouvelles substances [3] dont cinq constituent un progrès thérapeutique majeur. Malgré les augmentations des investissements en R & D, le nombre de médicaments définis comme réellement innovants est resté stable aux États-Unis, environ vingt par an selon la Food and Drug Administration (FDA) depuis 1994 [4] (Drew, 2000). Parallèlement, le coût de développement de ces nouveaux médicaments et leur prix ne cessent d’augmenter.
4Dans ce contexte, la question de l’innovation pharmaceutique est une préoccupation centrale pour le régulateur qui se trouve en effet tenu, d’une part, de soutenir la dynamique et la compétitivité des industriels et, d’autre part, de maîtriser la croissance des dépenses tout en préservant la qualité des soins et leur accessibilité. Comment dans ce cadre valoriser l’innovation, sans pour autant mettre en danger les capacités du système à financer des dépenses sans cesse croissantes ?
5En effet, si dans le cas général des biens industriels s’échangeant sur un marché concurrentiel, le soutien à l’innovation se limite à des aides publiques au financement de la recherche et après l’innovation à un cadre juridique de protection (brevets, licences), l’innovation pharmaceutique, parce que la dépense de médicament est socialisée, se situe dans un tout autre cadre. L’arrivée d’une innovation sur le marché pharmaceutique représente alors un enjeu en termes de dépenses publiques, d’autant plus que, le consommateur n’étant pas le payeur, le prix ne régule pas, ou mal, la demande et la loi du marché ne fonctionne pas. Le régulateur se trouve ainsi devant la nécessité d’introduire des modes de régulation qui passent par trois outils possibles, une action sur les prix, une action sur le remboursement ou une action directe sur la dépense.
Cet article vise à faire le point sur la manière dont différents systèmes de santé prennent en compte cette innovation. Nous avons ainsi essentiellement identifié des outils agissant sur les prix (les prix de référence) et sur le remboursement ou son niveau (le service médical rendu, l’évaluation économique). Sans toutefois viser l’exhaustivité, nous utilisons pour illustrer notre propos quelques exemples sélectionnés dans des pays prenant en compte explicitement l’innovation au sein des processus de régulation du médicament (cf. tableau 1). Ces dix pays ont été choisis de manière raisonnée de façon à représenter à la fois une diversité de solutions en matière de prise en compte de l’innovation et une diversité de modes de régulation des prix et des remboursements des médicaments.
Régulation du médicament et outils utilisés pour valoriser l’innovation dans les 10 pays étudiés
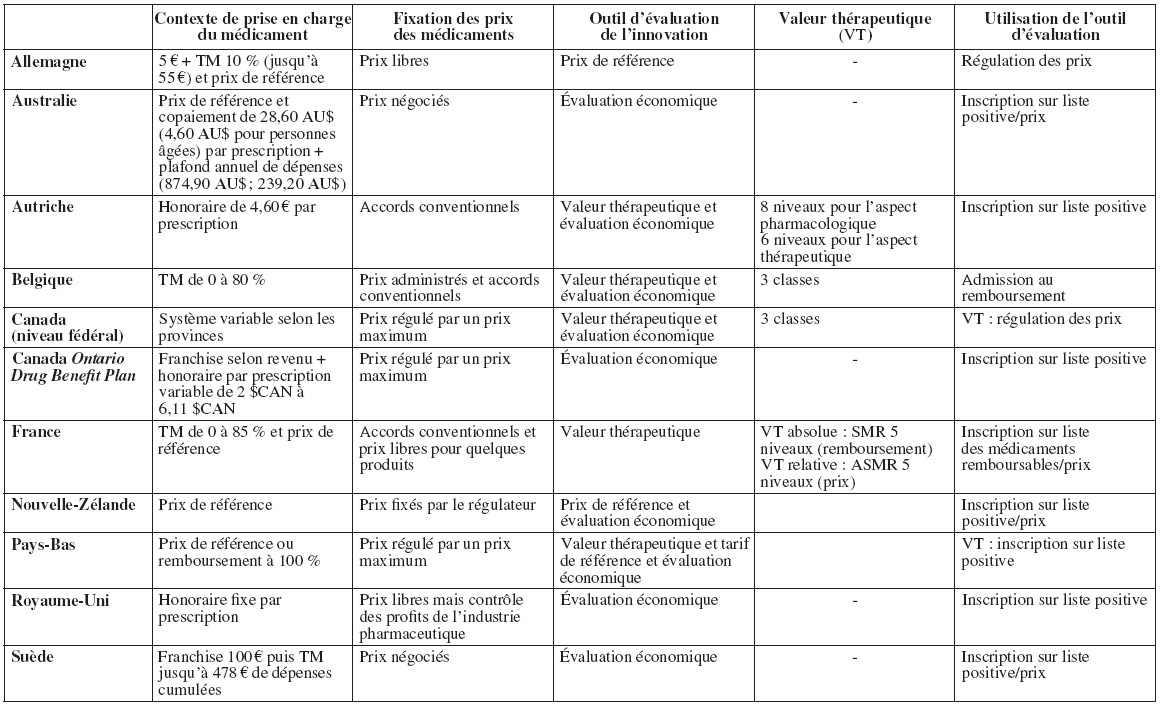
Régulation du médicament et outils utilisés pour valoriser l’innovation dans les 10 pays étudiés
Les outils
6La prise en compte du caractère plus ou moins innovant d’un médicament dans les politiques de fixation des prix ou de prise en charge publique met en œuvre un certain nombre d’outils très différents qui sont, selon les cas, utilisés séparément ou intégrés dans des processus plus complexes. Ces outils et la manière dont ils sont utilisés sont souvent révélateurs de la plus ou moins grande importance accordée par le système à l’innovation.
La « valeur thérapeutique ajoutée »
7Dans certains systèmes de santé c’est la « valeur thérapeutique ajoutée » du médicament qui est prise en compte. Il s’agit d’une évaluation relative visant à positionner un médicament par rapport à ceux qui existent dans la même classe thérapeutique. Cette notion de « valeur thérapeutique ajoutée » apparaît dans le discours européen au début des années 2000 [5]. Le Conseil européen souligne alors l’importance que revêt l’identification des médicaments présentant une forte valeur ajoutée thérapeutique en tant que facteur de promotion de l’innovation. Des textes ultérieurs préciseront que cette efficacité relative doit être prise en compte au moment de décider du remboursement du médicament et que cette « question… doit rester distincte du processus d’autorisation» [6], [7]. L’utilisation de cette « valeur thérapeutique ajoutée» est parfois largement antérieure à la législation européenne. Ainsi, la notion d’amélioration du service médical rendu (ASMR) a été définie en France pour la première fois en 1990. Elle n’est en revanche apparue qu’en 2001 dans la législation belge, sous la dénomination de valeur thérapeutique ajoutée ou plus-value.
8Les grilles servant à son évaluation sont variables suivant les pays. Elles prennent parfois une forme très synthétique, les différents critères étant résumés dans une appréciation globale, telle que l’« amélioration du service médical rendu » en France ou la « plus-value » en Belgique. Dans d’autres cas, comme en Autriche, il existe des grilles qui apprécient séparément l’innovation pharmaceutique et le bénéfice thérapeutique. Certaines sont très simplifiées et ne comportent que deux ou trois niveaux tandis que d’autres plus détaillées, apprécient jusqu’à huit niveaux différents.
9Suivant les pays, cette valeur thérapeutique conditionne soit l’admission au remboursement d’un produit ou le niveau de sa prise en charge, soit la fixation ou le plafonnement de son prix, le plus souvent les deux (Jacobzone, 2000) [8]. Elle peut être évaluée selon plusieurs critères et à des étapes différentes du processus de régulation dans laquelle elle se situe. Les exemples ci-après illustrent ces différents types d’utilisation.
• La France
10En France, l’admission au remboursement et la fixation des prix des médicaments sont soumises à deux évaluations complémentaires réalisées par la Commission de transparence à laquelle sont soumises les demandes d’inscription sur la liste des médicaments remboursables (Grandfils, 2007). Il s’agit du service médical rendu (SMR) [9] qui détermine le degré de prise en charge par l’assurance maladie et de l’amélioration du service médical rendu (ASMR) qui est le critère central entrant en jeu dans la fixation des prix.
11Le SMR est un critère absolu qui prend en compte la gravité de la pathologie pour laquelle le médicament est indiqué, l’efficacité et les effets indésirables, sa place dans la stratégie thérapeutique, notamment au regard des autres thérapies disponibles, et l’existence d’autres alternatives thérapeutiques, ainsi que son intérêt pour la santé publique. Plusieurs niveaux de SMR ont été définis : SMR majeur, important, modéré, faible et insuffi-sant. Le SMR est mesuré à un moment donné et peut évoluer dans le temps en fonction des modifications des données sur lequel il se fonde ou lorsque des alternatives plus efficaces apparaissent. Il est régulièrement réévalué lors du renouvellement d’inscription du médicament qui intervient tous les cinq ans. En combinaison avec la gravité de la maladie, le SMR détermine le niveau de remboursement (65 %, 30 %, ou 0 %) susceptible d’être accordé au médicament.
12L’ASMR est un critère relatif qui mesure le progrès thérapeutique ou la valeur ajoutée du médicament par rapport à des médicaments de la même classe thérapeutique. L’ASMR comporte cinq niveaux :
- l’ASMR I est attribué aux produits présentant un progrès thérapeutique majeur ;
- l’ASMR II désigne une amélioration importante en termes d’efficacité ou de réduction des effets indésirables ;
- l’ASMR III qualifie une amélioration modeste en termes d’efficacité ou de réduction des effets indésirables ;
- l’ASMR IV, une amélioration mineure en termes d’efficacité et/ou d’utilité ;
- et l’ASMR V, une absence d’amélioration avec avis favorable à l’inscription.
• Le Canada
13Au Canada, l’évaluation de la valeur ajoutée du médicament s’intègre dans un instrument de régulation des prix des médicaments dont l’objectif est de limiter les prix des médicaments encore sous brevet et de protéger les consommateurs contre des prix « excessifs » (Paris, Docteur, 2006). Cette régulation est sous la responsabilité du Patented Medicine Prices Review Board (PMPRB)/ Conseil d’examen du prix du médicament breveté (CEPMB). Le conseil compare le prix proposé par le laboratoire aux prix d’autres médicaments existant sur le marché canadien ou au prix du médicament dans sept pays (Allemagne, France, Italie, Suède, Suisse, Royaume-Uni et États-Unis). Il s’appuie pour cette comparaison sur un classement réalisé par le Human Drug Advisory Panel (HDAP)/Groupe consultatif sur les médicaments pour usage humain (les résultats sont publiés sur internet [10]) des produits en trois catégories [11] selon leur degré d’innovation :
- catégorie 1 : nouvelle présentation d’une forme existante d’un médicament, ou nouvelle forme pharmaceutique comparable [12] à la forme pharmaceutique existante ;
- catégorie 2 : nouveau médicament constituant une découverte ou une amélioration importante par rapport aux médicaments existants ; il peut s’agir soit d’une nouvelle entité chimique, soit d’une nouvelle forme pharmaceutique d’un médicament existant présentant une innovation importante ;
- catégorie 3 : nouveau médicament apportant une amélioration modeste ou minime par rapport aux médicaments existants ; il peut s’agir soit d’une nouvelle entité chimique, soit d’une nouvelle forme pharmaceutique d’un médicament existant mais le bénéfice thérapeutique apporté par cette nouvelle forme est modeste, voire nul.
• La Belgique
14Le troisième exemple est celui de la Belgique. Ce pays a également mis en œuvre une classification de la valeur ajoutée qui est utilisée dans le cadre de l’admission au remboursement du médicament. Toute demande de prise en charge de médicament est examinée par la Commission de remboursement du médicament constituée au sein de l’assurance maladie belge, l’Institut national d’assurance maladie-invalidité (INAMI). Cette commission détermine une valeur thérapeutique pour chaque produit, prenant en compte l’efficacité, l’utilité, les effets indésirables, l’applicabilité et le confort [13], [14]. La loi spécifie que cette évaluation doit tenir compte de la morbidité, de la mortalité ou de la qualité de vie et qu’une spécialité possède une plus value thérapeutique lorsque le traitement à l’aide de la spécialité donne lieu à une valeur supérieure à celle d’un traitement existant. Cette valeur thérapeutique est exprimée en trois classes, dites de « plus-value » :
- classe 1 : spécialités pharmaceutiques ayant une plus-value thérapeutique démontrée par rapport aux alternatives thérapeutiques existantes ;
- classe 2 : spécialités pharmaceutiques n’ayant pas une plus-value thérapeutique démontrée par rapport aux alternatives thérapeutiques existantes. Cette classe comporte trois sous-classes permettant notamment d’isoler les extensions de gamme d’une spécialité déjà remboursable ;
- classe 3 : cette dernière catégorie concerne essentiellement les génériques.
• L’Autriche
15Le dernier exemple que nous avons retenu est illustratif du degré de précision important qui a parfois été adopté dans certains pays. En Autriche, les médicaments ne sont remboursés que s’ils figurent dans la liste positive des biens remboursables (Erstattungcodex). L’évaluation du médicament est réalisée par une commission (Heilmittelevaluierungkommisson – HEK) qui émet un avis à destination de l’association des caisses d’assurance maladie qui prend la décision finale d’inscription. Trois évaluations sont conduites successivement : pharmacologique, médicale et économique.
16L’évaluation pharmacologique a deux objectifs : d’une part définir le degré d’innovation du médicament et d’autre part choisir les comparateurs. La classification du caractère innovant sur le plan pharmacologique se fait selon huit niveaux :
- même principe actif, même dosage et même voie d’administration qu’un médicament déjà inscrit sur la liste (générique) ;
- même principe actif, même voie d’administration, mais dosage différent ;
- nouvelle combinaison de principes actifs existants ;
- nouvelle voie d’administration d’un principe actif existant ou d’une combinaison de principes actifs existants ;
- nouveau principe actif d’une classe thérapeutique existante ;
- nouveau principe actif dans une nouvelle classe thérapeutique pour le traitement d’une maladie pour laquelle il existe déjà des médicaments inscrits dans la liste ;
- premier traitement médicamenteux d’une maladie pour laquelle il existait auparavant d’autres traitements non médicamenteux ;
- premier traitement d’une maladie qui ne pouvait pas être traitée auparavant.
- pas de bénéfice thérapeutique supplémentaire par rapport aux médicaments comparables ;
- même bénéfice ou un bénéfice similaire pour les patients et est donc considéré comme une nouvelle option thérapeutique ;
- bénéfice thérapeutique supplémentaire en comparaison des alternatives thérapeutiques pour un sous-groupe de patients ;
- bénéfice thérapeutique supplémentaire en comparaison des alternatives thérapeutiques pour la majorité des patients ;
- bénéfice thérapeutique supplémentaire important en comparaison des alternatives thérapeutiques pour un sous-groupe de patients ;
- bénéfice thérapeutique supplémentaire important en comparaison des alternatives thérapeutiques pour la majorité des patients.
Les prix de référence
17Même si les prix de référence ne sont pas explicitement conçus pour cela, le classement d’un médicament dans une catégorie soumise à prix de référence conduit le plus souvent à évaluer implicitement ou explicitement son degré d’innovation. Ce système consiste en effet à réunir des médicaments considérés comme comparables dans des groupes ou classes thérapeutiques auxquels on applique ensuite un tarif unique de prise en charge ou de remboursement. Les classes ainsi constituées peuvent être uniquement formées de médicaments génériques, avec le même principe actif et la même formulation. Elles peuvent aussi rassembler au sein de classes thérapeutiques plus ou moins larges, des principes actifs différents dont les indications sont proches. Certains pays incluent dans ces classes des médicaments encore sous brevet. Dans cette dernière configuration, et dans le cas de larges classes thérapeutiques, l’inclusion d’un nouveau médicament dans un groupe existant nécessite d’évaluer le degré d’innovation relatif de la nouvelle molécule.
• L’Allemagne
18L’Allemagne a été la première à mettre en place une politique de prix de référence, le Festbetrag, en 1989. Trois catégories de médicaments soumis à prix de référence ont progressivement été créées : la première regroupe les médicaments composés du même principe actif, la deuxième les médicaments composés de principe actifs chimiquement similaires et le troisième rassemble des médicaments dont seul l’effet thérapeutique est similaire. De 1989 à 1995 puis depuis 2004, les médicaments encore sous brevet sont également inclus dans ce système et des groupes de médicaments ont été créés, comportant à la fois des produits sous brevet et des génériques. Ces groupes sont appelés « jumbo groups ». Ainsi, toutes les molécules introduites sur le marché doivent être préalablement évaluées par le Gemeinsammer Bundesausschuss (GBA) qui décide si leur apport thérapeutique est suffi-sant pour qu’ils échappent au système de prix de référence. La loi de modernisation du système d’assurance maladie [17] (AVWG) précise les conditions nécessaires pour qu’un médicament apporte une amélioration thérapeutique : une utilité plus importante que les autres médicaments de son groupe ou une diminution de la fréquence ou de la sévérité des effets secondaires.
• Les Pays-Bas
19De la même manière aux Pays-Bas, le nombre de classes de médicaments soumis à prix de référence est très étendu : plus de 400 classes ont ainsi été définies et elles s’élargissent au fur et à mesure de l’expiration des brevets. Les classes sont basées sur la classification ATC [18] de l’OMS et chaque classe comporte au moins deux médicaments. Tous les médicaments qui sont substituables sont inclus dans cette liste, dite « annexe 1A», qui peut également comprendre des médicaments sous brevet. Les médicaments de cette liste sont remboursés sur la base d’un forfait déterminé pour chacune des classes. Si le médicament est plus onéreux, le patient paie la différence. En tout état de cause, il existe au moins un médicament remboursé à 100 % dans chacune des classes (Stolk, Rutten, 2007).
• La Nouvelle Zélande
20Une politique extensive de prix de référence existe également en Nouvelle-Zélande. Les médicaments sont d’abord regroupés en classes de médicaments traitant des pathologies identiques ou similaires puis en sous-classes de médicaments produisant un effet thérapeutique identique ou similaire. Les médicaments appartenant à la même sous classe sont pris en charge au prix du médicament le moins cher de la sous-classe, et ceci même dans le cas où les médicaments ne sont pas strictement substituables. Ainsi, le prix du médicament le moins cher définit le niveau de remboursement de toute la sous-classe sans différentiation en fonction de l’effet thérapeutique (Sundakov, Sundakov, 2007). Tout nouveau produit doit être systématiquement introduit dans une classe, faute de se voir refuser le remboursement. Les produits qui n’entrent pas dans une classe existante peuvent être remboursés si l’organisme régulateur (PHARMAC) et la firme s’accordent sur le prix du médicament. Ces critères très stricts génèrent des retards importants dans l’admission de nouveaux médicaments sur la liste positive. Serevent® a ainsi mis cinq ans à être listé. Imigran® a été soumis quatre fois en cinq ans (Danzon, Ketcham, 2003).
21Le système néo-zélandais, en poussant à l’extrême la mise en œuvre des prix de référence, est très révélateur de la nature exacte de l’innovation contenue dans les nouveaux produits. En effet, soit le médicament contient une innovation radicale et n’entrera dans aucune des classes existantes, il se verra alors attribuer un prix par l’instance régulatrice, soit il entre dans une classe existante et les innovations incrémentales dont il est porteur ne peuvent bénéficier d’une valorisation par les prix qu’en échange d’un abandon de l’admission sur la liste positive donc des produits remboursables.
22L’intérêt théorique du prix de référence réside dans le fait qu’il est supposé stimuler la compétition par les prix en augmentant l’élasticité de la demande,
et ceci, d’autant plus que les groupes sont plus larges (Brekke, Konigbauer, Straume, 2007). Les prix des médicaments appartenant au même groupe ou à des groupes proches pour la même indication varient alors en fonction de la valeur thérapeutique qu’ils apportent (Sheridan, Attridge, 2006). Les produits innovants qui ne sont pas inclus dans le système des prix de référence bénéficient alors de prix libres, plus ou moins contrôlés qui permettent aux laboratoires pharmaceutiques de valoriser leurs investissements. Toutefois ces politiques de prix de référence sont assez contestées et souvent accusées de freiner l’innovation pharmaceutique en diminuant les revenus que les laboratoires peuvent espérer de leurs innovations (Sheridan, Attridge, 2006).
Le rôle de l’évaluation économique
23Un certain nombre de pays ont choisi de fonder les décisions de prise en charge des innovations sur des évaluations économiques. Ces dernières permettent d’évaluer des technologies qui procurent un bénéfice additionnel mais à un supplément de coût. Grâce à ces méthodes, les nouvelles technologies ne sont prises en charge que si leur bénéfice, de préférence mesuré en QALYs [19], compense un certain niveau de coût (Jena, Philipson, 2007). Dans les milieux de l’industrie pharmaceutique, ces évaluations économiques sont désormais considérées comme étant la quatrième barrière « the fourth hurdle » à la commercialisation d’un médicament, les autres barrières étant l’efficacité, la sécurité et la qualité (Taylor, Drummond, Salkeld, Sullivan, 2004 ; Cohen, Cairns, Paquette, Faden, 2006).
24La place de l’évaluation économique varie selon le système de santé. Elle peut faire partie des critères pour l’inclusion des médicaments dans les listes positives, dans des formulaires des régimes d’assurances ou des recommandations de prise en charge. C’est le cas dans des pays dont le système de fixation des prix des médicaments est libre comme au Royaume-Uni (Pearson, Rawlins, 2005). Ailleurs, elle peut être aussi un des critères utilisés dans les processus de régulation des prix. Ainsi, la province de l’Ontario au Canada a été l’une des premières à utiliser les résultats de l’évaluation économique dans le processus de décision de remboursement (Laupacis, 2005). En Australie, par exemple, elle est à la fois utilisée par le Pharmaceutical Benefits Advisory Committee (PBAC) pour l’inclusion dans la liste positive, et par la Pharmaceutical Benefit Pricing Authority (PBPA) pour la fixation du prix (Henry, Hill, Harris, 2005 ; Faunce, 2007). En Suède, c’est un critère prépondérant et le laboratoire doit proposer un prix qui soit coût-efficace pour que le remboursement soit accepté par le Läkemedelsförmånsnämnden (LFN) (Moïse, Docteur, 2007). Dans les systèmes avec prix de référence, l’évaluation économique pourrait être utilisée lorsque les prix de référence s’étendent à des médicaments similaires, voire d’action similaire, comme en Allemagne. Dans ces cas, l’équivalence ou non de deux produits serait estimée par l’évaluation économique. Le cas du Sumatriptan, nouvel antimigraineux, initialement classé aux Pays-Bas dans le même groupe de référence que les autres antimigraineux de type ergotamine, et ce malgré les affirmations du laboratoire sur sa meilleure efficience, est révélateur de l’utilité qu’aurait pu revêtir l’évaluation économique dans un pays utilisant les prix de référence (Drummond, Jonsson, Rutten, 1997). Dans certains pays et en particulier la France, le Danemark ou l’Italie, l’évaluation économique est facultative (Taylor et al., 2004).
25Il existe différentes formes d’évaluation, mais la plupart des pays préconisent de réaliser des études « coût-utilité » et d’évaluer ainsi les coûts supplémentaires par QALYs gagnés grâce à la nouvelle technologie (Ontario Ministry of Health and Long-Term Care, 2007 ; Pearson, Rawlins, 2005 ; Henry et al., 2005). Lorsque la mesure des QALYs n’est pas nécessaire ou pas possible, d’autres types d’évaluation tels que les analyses coût-efficacité, coût-bénéfice ou les études de minimisation des coûts sont aussi acceptés. D’autres indicateurs sont alors utilisés tels que le nombre de problèmes de santé évités, le nombre d’effets secondaires évités, le coût par année de vie gagnée. D’une manière générale, les critères pris en compte pour évaluer l’efficacité diffèrent d’un système à un autre et ils ne sont pas sans incidence sur la reconnaissance de l’innovation, ainsi que pour la prise en compte du confort du patient.
26Un des principes fondamentaux de l’évaluation économique repose sur l’idée que seuls les médicaments « coûts-efficaces » sont susceptibles d’être recommandés tandis que ceux qui ne le sont pas sont exclus du remboursement. Il est donc légitime de s’interroger sur le niveau de coût acceptable et le seuil au-delà duquel un médicament doit être rejeté. Une étude, réalisée en Australie, en Belgique et au Royaume-Uni, montre qu’aucun de ces trois pays n’affiche explicitement un seuil (Henry et al., 2005 ; Laupacis, 2005). Toutefois, le NICE au Royaume-Uni est arrivé de manière opérationnelle à une fourchette de 30 600 dollars à 45 900 dollars (1 dollar = 0,65 livre) au-delà de laquelle le risque de rejet augmente (Pearson, Rawlins, 2005). De son côté, le PBAC en Australie n’a accepté aucun médicament au-dessus du seuil de 52 400 dollars entre 1994 et 2003. Toujours en Australie, George et al. ont analysé 72 demandes d’admission au remboursement. 36 d’entre elles avaient un coût par QALYs supérieur à 57 000 dollars et seules deux ont été acceptées. À l’inverse, il n’y a eu qu’un seul rejet pour les 26 soumissions dont le coût était inférieur à 32 000 dollars (George, Harris, Mitchell, 2001). De son côté, la Belgique n’a pas non plus de seuil offi-ciel, mais le niveau de 40000 dollars par QALYs est généralement considéré comme un maximum (Adriaens, Antonissen, Arickx, de Haes, Van de Velde, Van Wilder, 2007). De ces analyses on peut conclure qu’il n’existe pas de seuil préalablement fixé et que d’autres facteurs peuvent être pris en compte comme par exemple l’impact sur le budget, la gravité de la maladie, l’existence d’autres traitements, voire même l’impact sur le style de vie.
Selon les cas, les recommandations s’appliquent sur un ensemble de médicaments ou sur une molécule en particulier. Ainsi, le NICE au Royaume-Uni émet des recommandations sur des classes thérapeutiques entières (exemple, l’ensemble de la classe des glitazones, médicament pour le diabète) tandis que le PBAC en Australie statue sur le prix et sur le coût total de présentations pharmaceutiques bien précises (rosiglitazone, pioglitazone, etc.) (Henry et al., 2005).
La mise en place de telles évaluations économiques conduit à rejeter une proportion très variable de demandes d’admission. En Suède, pendant les trente premiers mois d’activité du LFN, d’octobre 2002 à mars 2005, sur 107 demandes de remboursement, 82 ont été acceptées sans restriction, 12 ont été acceptées sous conditions et 13 ont été refusées (12 %) (Anell, Persson, 2005). Dans le travail de George et al. en Australie, le pourcentage de demandes refusées était de 37 % (George et al., 2001). En Ontario, premier État canadien à avoir institué les évaluations économiques préalablement à l’admission au remboursement, 58 % des médicaments examinés sur la période 2000-2001 ont été rejetés lors de leur première soumission. Ces écarts entre pays s’expliquent probablement par le poids variable accordé à l’évaluation économique parmi tous les critères conduisant à la décision.
Les procédures
27L’estimation de la valeur thérapeutique, l’évaluation économique ou la détermination de groupes de produits soumis à prix de référence s’intègrent à différentes étapes dans les processus de régulation du médicament.
Procédures centralisées d ’évaluation clinique et économique
28L’évaluation du médicament peut ainsi s’inscrire au sein de procédures centralisées d’évaluation clinique et économique des médicaments, les Centralized Drug Review (CDR). De telles procédures ont été mises en place par exemple en Australie, au Canada, en Nouvelle-Zélande ou au Royaume-Uni. Leur caractère national et centralisé facilite l’évaluation comparative et la priorisation des produits pharmaceutiques. D’une manière générale, le travail réalisé par ces CDR se fait en deux étapes.
29La première étape est une évaluation clinique et économique basée sur une revue systématique des études cliniques et une analyse critique des études pharmaco-économiques adressées par les firmes. Elle vise à mesurer l’impact du médicament sur la santé des patients et sur le système de santé. C’est une étape essentiellement scientifique.
30L’étape suivante est celle des recommandations sur le remboursement ou la prise en charge. Il s’agit ici de confronter l’évidence clinique et économique aux priorités de chaque système de santé. La question est de savoir si le médicament doit être pris en charge et dans quelles conditions (Morgan, McMahon, Mitton, Roughead, Kirk, Kanavos et al., 2006 ; McMahon, Morgan, Mitton, 2006).
31Les différences principales entre ces procédures d’évaluation du médicament résident dans leur degré d’intégration dans les processus de prise en charge ou de remboursement. En Australie, comme en Nouvelle-Zélande, ou encore en Suède, elles aboutissent directement à des décisions de prise en charge. En Australie, le PBAC fait directement ses recommandations au ministre de la Santé en vue de l’admission des médicaments sur la liste de prise en charge par le National Pharmaceutical Benefit Scheme (PBS). Le ministre ne prend aucune décision d’inscription sans une recommandation du PBAC. En Nouvelle-Zélande, c’est une agence nationale, PHARMAC (Pharmaceutical Management Agency), qui gère l’inscription sur la liste positive de même que le prix du médicament. En Suède, le LFN décide du remboursement de tous les médicaments disponibles à la prescription. Après négociation avec le laboratoire il en fixe également le prix.
Ailleurs, l’évaluation économique est réalisée par des organisations indépendantes des instances décisionnaires et les recommandations ainsi émises peuvent ou non s’imposer au financeur. Au Royaume-Uni, le National Institute for Clinical Excellence (NICE) réalise des recommandations de prise en charge à destination du National Health Service (NHS) d’Angleterre et du pays de Galles. Ces recommandations sont obligatoirement suivies par le NHS. Cependant, le NICE n’évalue qu’une petite partie des médicaments sur le marché et il reste d’importantes disparités de prise en charge pour tous les autres médicaments. Au Canada (voir encadré 1), l’évaluation économique des nouveaux produits pharmaceutiques est réalisée par deux entités différentes. Initialement réalisées au niveau local dans certaines provinces comme l’Ontario, il existe désormais une procédure centralisée pour les nouvelles molécules au sein de l’Agence canadienne des médicaments et des technologies de la santé/Canadian Agency for Drug and Technology in Health (ACMTS/ CADTH). L’objectif final est de produire des recommandations sur l’inclusion ou non des médicaments dans les formulaires des régimes publics d’assurance médicament du Canada, à l’exception du Québec. Ces recommandations ne sont pas obligatoirement suivies par les régimes d’assurance médicament.
Encadré 1 : Un exemple de processus de régulation du médicament : le Canada
- vérifie efficacité, sécurité et qualité ;
- évalue le bénéfice risque ;
- réalise une surveillance post-marketing.
3) Canadian Agency for Drugs and Technology in Health (CADTH)/ Agence canadienne des médicaments et technologies de la santé (ACMTS) – améliore l’accès et l’utilisation d’une information basée sur les preuves – organise les CDR qui évaluent les nouveaux médicaments dans l’optique de leur remboursement par les régimes d’assurance médicament (fédéraux, provinciaux ou territoriaux).
Le CADTH réalise des évaluations cliniques et pharmaco-économiques pour estimer le coût-efficacité du médicament. Ces CDR sont utilisés par le Canadian Expert Drug Advisory Committee (CEDAC)/ Comité consultatif canadien d’expertise sur les médicaments (CCEM) comme base pour les recommandations d’inclusion ou non des médicaments dans les formulaires.
Les gouvernements fédéraux, provinciaux ou territoriaux examinent les recommandations du CDR et prennent la décision finale.
4) Évaluation du médicament au niveau local qui – décide de l’inclusion des médicaments dans la liste positive – évalue la substituabilité générique/médicament sous brevet ; en cas de substitution possible fixe le tarif de remboursement au niveau du générique – évalue la valeur thérapeutique et le coût-efficacité du nouveau médicament pour le compte des régimes d’assurance médicaments à partir des analyses effectuées par le CADTH – évalue l’impact sur les dépenses publiques de l’introduction d’un nouveau médicament dans la liste positive.
Absence d ’évaluation centralisée de type CDR
32D’autres pays ne pratiquent pas une évaluation centralisée, telle qu’elle est contenue dans le concept de CDR (Centralized Drug Review). Les différents outils d’évaluation de l’innovation du médicament sont alors utilisés dans l’une ou l’autre des étapes du processus de régulation.
33Ainsi, en Allemagne, l’inclusion dans un groupe de prix de référence est réalisée par le Gemeinsammer Bundes Ausschuss (GBA), le Comité fédéral conjoint, organisme fédérateur des professions médicales et des caisses d’assurance maladie. L’évaluation économique jusqu’ici peu pratiquée a été confiée à une agence publique nouvellement créée, l’Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Toutefois depuis sa création, l’IQWiG a davantage réalisé des évaluations de l’efficacité des médicaments et produit des recommandations de prescriptions à l’usage des médecins (Garattini, Cornago, De Compadri, 2007). En France, la valeur thérapeutique du médicament est attribuée par la Commission de transparence, puis elle est utilisée par le Comité économique des produits de santé (CEPS) pour la fixation du prix. Aucune évaluation économique n’était requise jusqu’à présent et les informations économiques demandées se limitent à la population cible, les médicaments comparateurs et à l’impact budgétaire (Grandfils, 2007).
34Dans tous les cas, le caractère innovant du médicament ou sa valeur ajoutée ne sont qu’un des critères pris en compte dans la décision. En Suède, par exemple, le LFN prend en compte trois principes fondamentaux : le principe de la « dignité humaine » selon lequel les soins de santé doivent être prodigués de manière équitable pour tous, le principe du besoin et de la solidarité qui assure les soins à ceux qui ont les besoins les plus grands, le principe de coût efficacité selon lequel le coût d’un médicament doit être raisonnable d’un point de vue médical, humain et économique et social et le principe de l’utilité marginale, partie du principe de coût efficacité (Moïse, Docteur, 2007 ; Anell, Persson, 2005). En Australie, le PBAC prend en compte dans sa décision l’efficacité et la sécurité du médicament, les avantages thérapeutiques et les inconvénients, le coût efficacité ainsi que la place relative du médicament par rapport aux thérapeutiques déjà prises en charge (Mitton, McMahon, Morgan, Gibson, 2006).
35Aux Pays-Bas, valeur thérapeutique, prix de référence et évaluation économique sont utilisées à des niveaux différents de la procédure de remboursement (cf. figure 1). Les prix des médicaments sont fixés par les firmes pharmaceutiques, dans les limites d’un prix maximum calculé en faisant la moyenne des prix dans quatre pays européens : la Belgique, la France, l’Allemagne et le Royaume-Uni. Parallèlement, le médicament doit entamer une procédure d’inscription dans une des listes positives : l’annexe 1A pour les médicaments soumis à prix de référence et l’annexe 1B pour les autres. Cette procédure est sous la responsabilité du ministre de la Santé guidé par les recommandations du Pharmaceutical Care Committee.
L’inclusion dans la liste des produits soumis à prix de référence (1A) fait en premier lieu appel à la notion d’interchangeabilité de la molécule : si sur le plan thérapeutique, la molécule est substituable par une autre, alors elle sera inscrite sur cette liste. Les autres médicaments, sont inclus dans la liste, dite « schedule 1B » des médicaments remboursés au prix proposé par le fabricant. Pour ces médicaments, le fabricant doit fournir une information sur la valeur thérapeutique et le coût efficacité du médicament. Ces éléments seront examinés par le comité qui détermine la valeur thérapeutique du produit en prenant en compte différents critères : l’effet thérapeutique, l’efficacité, les effets secondaires, la population cible, la facilité d’utilisation, l’amélioration de la qualité de vie du patient et l’impact budgétaire (Garattini et al., 2007 ; Pronk, Bonsel, 2004 ; Stolk, Rutten, 2007).
Schéma des décisions de prise en charge du médicament au Pays-Bas

Schéma des décisions de prise en charge du médicament au Pays-Bas
36Les critiques émises à l’encontre de l’évaluation économique sont nombreuses. Elles proviennent des patients qui y voient une restriction à l’accès aux médicaments, des firmes pharmaceutiques qui considèrent qu’elle étouffe l’innovation, des médecins qui l’accusent de limiter le champ de leur pratique, voire même des décideurs politiques qui estiment qu’elle encourage l’augmentation des dépenses de santé (Laupacis, 2005). Ainsi, on reproche en particulier aux évaluations économiques de freiner l’entrée sur le marché des médicaments innovants (Lundkvist, Jonsson, Rehnberg, 2006). Le NICE par exemple est soupçonné d’avoir un impact négatif sur la rapidité de prise en charge des médicaments, car les Primary Care Trust du NHS retardent leurs décisions dans l’attente de ses recommandations (Cohen et al., 2006). Au Canada, l’industrie pharmaceutique reproche aux autorités de ne pas évaluer correctement les bénéfices des nouveaux médicaments et de ne pas avoir su accorder de prime à des nouveaux produits améliorant le confort des patients (Paris, Docteur, 2006).
37Dans un autre registre, Jena et Philipson considèrent que si l’évaluation économique est positive pour le bien-être des patients actuels, elle est négative pour les patients futurs, car en limitant les profits de l’industrie pharmaceutique, elle freine en même temps l’innovation (Jena, Philipson, 2007). L’absence de transparence des organismes régulateurs génère également des doutes sur l’objectivité des décisions qui sont prises (Laupacis, 2005). Parallèlement, les études pharmaco-économiques elles-mêmes sont parfois sévèrement critiquées et soupçonnées de partialité, en particulier lorsque leurs sponsors appartiennent à l’industrie pharmaceutique (Cornago, Li Bassil, De Compadri, Garattini, 2007).
Quelques exemples de produits et de leur évaluation selon les pays
38Les évaluations des médicaments et de leur degré d’innovation n’aboutissent pas toujours à des résultats comparables en termes de prise en charge. Morgan et al. ont effectué en 2003 une comparaison sur quatre pays (Australie, Canada, Nouvelle-Zélande, Royaume-Uni) de 17 médicaments parmi les plus vendus dans le monde (Morgan, 2006). Ils montrent que les décisions d’inclusion sur les listes positives sont loin d’être concordantes entre pays. Ceci reflète les différences d’interprétation des données sur le médicament, les disparités de prise en charge des alternatives thérapeutiques ainsi que le pouvoir de négociation des firmes pharmaceutiques. L’Australie et la Nouvelle-Zélande qui réalisent des évaluations très complètes de tous les produits, aboutissent finalement à une restriction de la liste des médicaments plus importante et à une moindre utilisation de ces médicaments.
39Nous avons de notre côté examiné la situation au regard de la prise en charge de quelques médicaments récemment arrivés sur le marché (cf. tableau 2). Nous avons retenu dans cette liste une dizaine de médicaments représentant des situations diverses : des médicaments très onéreux, des médicaments avec une valeur thérapeutique importante ou non, des médicaments comportant des innovations majeures ou encore des « me-too ». Sur ce petit échantillon, nous arrivons à une conclusion similaire. Il semble effectivement que les pays où sont réalisées des évaluations plus complètes, incluant des évaluations économiques, sont aussi des pays où le remboursement ou la prise en charge des médicaments est plus restreinte. L’Australie, la Nouvelle-Zélande et le Canada sont dans ce cas. En revanche, les critères de la Suède paraissent moins restrictifs, bien que ce pays intègre également une évaluation économique dans le processus.
Encadré 2 : Un exemple de variabilité de prise en charge : l’adéfovir dipivoxil
Ce même produit est entièrement pris en charge en Allemagne, où il n’a pas été intégré dans un groupe de prix de référence.
À l’inverse, dans deux pays où des études médico-économiques sont demandées à l’appui de la décision de prise en charge, la demande a été rejetée. La recommandation finale du Comité consultatif canadien d’expertise sur les médicaments est de ne pas inscrire l’adéfovir dipivoxil sur la liste des médicaments couverts, arguant de l’absence de démonstration de supériorité, en termes d’incidence des décès et de mortalité grave, par rapport aux médicaments comparables ainsi que d’un coût élevé non justifié par le rapport coût efficacité supplémentaire. On constate effectivement que ce médicament ne figure pas sur le formulaire de l’Ontario Drug Benefit Plan. En Australie, le PBAC a rejeté la première soumission du laboratoire en s’appuyant sur le manque d’information permettant de conclure à la supériorité du traitement en termes de coût efficacité. L’adéfovir dipivoxil n’a pu être admis sur la liste positive que plus tard.
Pour ce même produit le NICE [20] a considéré que les données étaient suffisantes pour que le produit soit pris en charge par le NHS sous certaines conditions. L’adéfovir dipivoxil est recommandé en cas d’inefficacité primaire ou secondaire du traitement par interféron alpha ou d’effets secondaires du traitement par l’interféron alpha. Par ailleurs, l’adéfovir dipivoxil n’est recommandé que si la personne a eu un traitement au préalable par la lamivudine, en cas de résistance à la lamivudine.
Inscription sur les listes positives ou présence sur le marché national de quelques produits récemment mis sur le marché selon les pays

Inscription sur les listes positives ou présence sur le marché national de quelques produits récemment mis sur le marché selon les pays
Conclusion
40Ce rapide tour d’horizon nous permet de mettre en évidence l’existence d’un certain nombre d’outils de valorisation de l’innovation utilisés par les différents systèmes de santé lors de la fixation du prix ou l’admission au remboursement d’un médicament. L’évaluation de la valeur thérapeutique ajoutée par le nouveau médicament, comparativement à ceux qui existent déjà sur le marché est utilisée dans beaucoup de pays. Elle y revêt des formes variées, utilisant des échelles plus ou moins précises et intervenant seule ou au sein de procédures plus élaborées. Les politiques de prix de référence, lorsqu’elles sont élargies aux médicaments sous brevet, constituent également une autre forme d’évaluation de l’innovation. Elles sont relativement peu répandues, en tout cas dans cette forme extensive. Les politiques de majorations de prix ne sont pas développées et ne sont pas appliquées dans le seul pays où elles sont inscrites dans la réglementation.
41Enfin, l’évaluation économique constitue la forme la plus élaborée et la plus aboutie de prise en compte de l’innovation. Au cours des processus qui conduisent à cette évaluation économique l’ensemble des aspects cliniques, populationnels et économiques sont pris en compte. Ce n’est pas seulement la valeur thérapeutique ajoutée du médicament qui entre dans les critères d’évaluation, mais aussi le différentiel de sécurité, d’efficacité, l’amélioration de la qualité de vie du patient, l’impact sur la mortalité et les coûts pour le système de santé. Cette évaluation économique est de plus en plus souvent au cœur du processus d’évaluation du médicament.
42L’utilisation qui est faite de ces différents outils a bien entendu un impact sur la liste des médicaments pris en charge et sur leur prix. Le premier constat est que ces procédures conduisent à des listes plus ou moins restrictives, les plus limitées semblant être celles de pays qui utilisent l’évaluation économique de manière très marquée. En termes d’utilisation, les médicaments qui ne figurent pas dans les listes positives ne sont que peu ou pas utilisés dans les pays concernés. Le second, assez fréquent, est celui de la lenteur de ces procédures qui retarderaient la mise à disposition de produits parfois importants et innovants. Enfin, on accuse ces procédures d’écarter de la prise en charge des innovations dont le seul bénéfice pour le patient se traduit en termes de confort.
Références
- ADRIAENS C., ANTONISSEN Y., ARICKX F., DE HAES J., VAN DE VELDE V., VAN WILDER P., (2007), Pharmaceutical Pricing and Reimbursement Information (PPRI), Pharma Profile : Belgium, PPRI Pharma Profile.
- ANELL A., PERSSON U., (2005), « Reimbursement and clinical guidance for pharmaceuticals in Sweden : do health-economic evaluations support decision making ? », Eur. J. Health Econ., 6 (3), 274-279.
- BREKKE K.R., KONIGBAUER I., STRAUME O.R., (2007), « Reference pricing of pharmaceuticals », J. Health Econ., 26 (3), 613-642.
- COHEN J., CAIRNS C., PAQUETTE C., FADEN L., (2006), « Comparing patient access to pharmaceuticals in the UK and US », Appl. Health Econ. Health Policy, 5 (3), 177-187.
- CORNAGO D., LI BASSIL L., DE COMPADRI P., GARATTINI L., (2007), « Pharmacoeconomic studies in Italy : a critical review of the literature », Eur. J. Health Econ., 8 (2), 89-95.
- DANZON P.M., KETCHAM J. D., (2003), Reference pricing of pharmaceuticals for medicare : evidence from Germany, the Nederlands and New Zeland, Cambridge : NBER.
- DRUMMOND M., JONSSON B., RUTTEN F., (1997), « The role of economic evaluation in the pricing and reimbursement of medicines », Health Policy, 40 (3), 199-215.
- FAUNCE T.A., (2007), « Reference pricing for pharmaceuticals : is the Australia-United States Free Trade Agreement affecting Australia’s Pharmaceutical Benefits Scheme ? », Med. J. Aust., 187 (4), 240-242.
- GARATTINI L., CORNAGO D., DE COMPADRI P., (2007), « Pricing and reimbursement of in-patent drugs in seven European countries : A comparative analysis », Health Policy, 82 (3), 330-339.
- GEORGE B., HARRIS A., MITCHELL A., (2001), « Cost-effectiveness analysis and the consistency of decision making : evidence from pharmaceutical reimbursement in australia (1991 to 1996) », Pharmacoeconomics, 19 (11), 1103-1109.
- GRANDFILS N., (2007), « Fixation et régulation des prix des médicaments en France », Revue française des Affaires sociales (n° 3-4 juin-décembre).
- HEALTH CANADA S.C., Access to Therapeutic Products : The Regulatory Process in Canada. http://www.hc-sc.gc.ca/ahc-asc/pubs/hpfb-dgpsa/access-therapeutic_accestherapeutique_e.html.2007.23-9-2007.
- HENRY D.A., HILL S.R., HARRIS A., (2005), « Drug prices and value for money : the Australian Pharmaceutical Benefits Scheme », JAMA, 294 (20), 2630-2632.
- JENA A.B., PHILIPSON T., (2007), « Cost-effectiveness as a price control », Health Aff. (Millwood.), 26 (3), 696-703.
- LAUPACIS A., (2005), « Incorporating economic evaluations into decision-making : the Ontario experience », Med. Care, 43 (7 Suppl), 15-19.
- LUNDKVIST J., JONSSON B., REHNBERG C., (2006), « The costs and benefits of regulations for reimbursement of new drugs », Health Policy, 79 (2-3), 337-344.
- MCMAHON M., MORGAN S., MITTON C., (2006), «The Common Drug Review : a NICE start for Canada ? », Health Policy, 77 (3), 339-351.
- MITTON C.R., MCMAHON M., MORGAN S., GIBSON J., (2006), « Centralized drug review processes : are they fair ? », Soc. Sci. Med., 63 (1), 200-211.
- MOÏSE P., DOCTEUR E., (2007), « Pharmaceutical Pricing and reimbursment policies in Sweden», OCDE Health Working Paper, OCDE, 28, 26 juillet 2007,
- MORGAN S.G., MCMAHON M., MITTON C., ROUGHEAD E., KIRK R., KANAVOS P., MENON D., (2006), « Centralized drug review processes in Australia, Canada, New Zealand, and the United kingdom », Health Aff. (Millwood.), 25 (2), 337-347.
- ONTARIO MINISTRY OF HEALTH AND LONG-TERM CARE, (2007), Ontario guidelines for Economic Analysis of Pharmaceutical Products, Ontario.
- PARIS V., DOCTEUR E., (2006), « Pharmaceutical Pricing and reimbursment policies in Canada », OCDE HEalth Working Paper, OCDE, 24, 22 décembre 2006.
- PEARSON S.D., RAWLINS M.D., (2005), « Quality, innovation, and value for money : NICE and the British National Health Service », JAMA, 294 (20), 2618-2622.
- PRONK M.H., BONSEL G.J., (2004), « Out-patient drug policy by clinical assessment rather than financial constraints ? The gate-keeping function of the out-patient drug reimbursement system in The Netherlands », Eur. J. Health Econ., 5 (3), 274-277.
- SHERIDAN D., ATTRIDGE J., (2006), « The Impact of Therapeutic Reference Pricing on Innovation in Cardiovascular Medicine », Pharmacoeconomics, 24 (suppl. 2), 35-54.
- STOLK E.A., RUTTEN F.F.H., (2007), The health basket in the Netherlands, Erasmus MC.
- SUNDAKOV A., SUNDAKOV V., (2007), New Zealand Pharmaceutical Policies, Castaglia Strategic Advisor.
- TAYLOR R.S., DRUMMOND M.F., SALKELD G., SULLIVAN S.D., (2004), « Inclusion of cost effectiveness in licensing requirements of new drugs : the fourth hurdle », BMJ, 329 (7472), 972-975.
- ZENTNER A., VELASCO-GARRIDO M., BUSSE R., (2007), Methoden zur vergleichenden Bewertung pharmazeutischer Produckte, DAHTA027, 147. 2007. Rüther A. HTA-Bericht.